Articles
- Page Path
- HOME > Ann Occup Environ Med > Volume 33; 2021 > Article
- Research Article Association between prenatal polycyclic aromatic hydrocarbons and infantile allergic diseases modified by maternal glutathione S-transferase polymorphisms: results from the MOCEH birth cohort
-
Tai Kyung Koh1,2
 , Hyesook Park2,3, Yun-Chul Hong4, Mina Ha5, Yangho Kim6, Bo-Eun Lee7, Surabhi Shah8, Eunhee Ha2,8
, Hyesook Park2,3, Yun-Chul Hong4, Mina Ha5, Yangho Kim6, Bo-Eun Lee7, Surabhi Shah8, Eunhee Ha2,8 -
Annals of Occupational and Environmental Medicine 2021;33:e12.
DOI: https://doi.org/10.35371/aoem.2021.33.e12
Published online: April 23, 2021
1Department of Occupational and Environmental Medicine, Ewha Womans University Mokdong Hospital, Seoul, Korea.
2Graduate Program in System Health Science and Engineering, Ewha Womans University, Seoul, Korea.
3Department of Preventive Medicine, College of Medicine, Ewha Womans University, Seoul, Korea.
4Department of Preventive Medicine, College of Medicine, Seoul National University, Seoul, Korea.
5Department of Preventive Medicine, Dankook University College of Medicine, Cheonan, Korea.
6Department of Occupational and Environmental Medicine, Ulsan University Hospital, University of Ulsan College of Medicine, Ulsan, Korea.
7Environmental Health Research Department, National Institute of Environmental Research, Incheon, Korea.
8Department of Occupational and Environmental Medicine, College of Medicine, Ewha Womans University, Seoul, Korea.
- Correspondence: Surabhi Shah. Department of Occupational and Environmental Medicine, College of Medicine, Ewha Womans University, 25 Magokdong-ro 2-gil, Gangseo-gu, Seoul 07804, Korea. surabhi.3007@gmail.com
- Correspondence: Eunhee Ha. Department of Occupational and Environmental Medicine, College of Medicine, Ewha Womans University, 25 Magokdong-ro 2-gil, Gangseo-gu, Seoul 07804, Korea. eunheeha@ewha.ac.kr
- *Equal contribution as co-corresponding authors.
• Received: May 21, 2020 • Accepted: April 5, 2021
Copyright © 2021 Korean Society of Occupational & Environmental Medicine
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background Prenatal exposure to polycyclic aromatic hydrocarbons (PAH) has been linked to allergic disease onset. Variations in the glutathione S-transferase (GST) gene family can impact the progression of allergic diseases. We sought to examine the association between prenatal PAH exposure and infantile allergic diseases in 6-month-old infants, and how maternal glutathione S-transferase M1 (GSTM1) or T1 (GSTT1) polymorphism affects the association between prenatal PAH exposure and allergic diseases in the Mothers and Children's Environmental Health (MOCEH) study.
-
Methods The study sample comprised 349 infants and their mothers from the MOCEH study, for whom 1-hydroxypyrene (1-OHP) and 2-naphthol were measured in both the early period of pregnancy and late period of pregnancy. An infant was deemed to be affected by an allergic disease if diagnosed with or if developed at least one of the allergic diseases. A logistic regression analysis was performed to study the association between urinary 1-OHP and 2-naphthol levels during pregnancy and allergic diseases in 6-month-old infants. Furthermore, analyses stratified by maternal GSTM1 or GSTT1 present/null polymorphisms were performed.
-
Results The risk of allergic diseases in 6-month-old infants was significantly increased in accordance with an increase in urinary 1-OHP during the early period of pregnancy (odds ratio [OR]: 1.84; 95% confidence interval [CI]: 1.05, 3.23; by one log-transformed unit of 1-OHP μg/g creatinine). The increased risk of infantile allergic diseases associated with urinary 1-OHP during the early period of pregnancy was limited to the maternal GSTT1 null type (OR: 2.69; 95% CI: 1.17, 6.21, by one log-transformed unit of 1-OHP μg/g creatinine); however, the Relative Excess Risk due to Interaction was not statistically significant.
-
Conclusions The present study found that infantile allergic diseases could be affected by intrauterine PAH exposure, particularly in the early prenatal period and the risk was limited to the maternal GSTT1 null type.
BACKGROUND
Over the last 2 decades, there has been an increase in the prevalence of childhood allergic diseases such as asthma, atopic dermatitis, food allergies, and allergic rhinitis. Allergic diseases are among the most common chronic diseases in both children and adults [1]. Based on the 2007 National Survey of Children's Health, the prevalence of atopic dermatitis in US children was 13.0% [2]. According to data from multicenter international studies, geographical variations of the prevalence of asthma range between 1.0% and 18.0%. Moreover, although epidemiologic studies on rhinitis are scarce, a nearly 30-fold increase (i.e., from 1.4% to 39.7%) was observed in the prevalence of rhinitis among 13–14-year-old children across 56 countries [3]. In the Republic of Korea, the prevalence of atopic dermatitis, asthma, and allergic rhinitis in 2014 was 19.0, 36.3, and 133.1 per 1,000 people, respectively [4], and the prevalence of atopic dermatitis in 6–7-year-old children increased from 8.8% in 1995 to 12.7% in 2015 [5].
According to “a study on research methodology and long-term planning regarding estimation of economic burden of major diseases in Korea (2009),” asthma was ranked fifth by occurrence among all examined chronic diseases [6]. The “developmental origins of health and disease” hypothesis proposes that in utero and early childhood exposure to environmental pollutants may increase susceptibility to allergic diseases [7]. Allergic diseases that are not properly managed in childhood and adolescence may become severe diseases in adulthood, thereby increasing economic burden and losses. Several risk factors are known to be associated with allergic diseases in epidemiologic studies, including early-life sensitization, traffic-related air pollution (black carbon, SO2, NO2, PM2.5, PM10, ozone), pre- or postnatal tobacco smoke exposure, viral infections (human rhinovirus, respiratory syncytial virus), mold, microbial exposure, and pets [8].
Polycyclic aromatic hydrocarbons (PAH) are fused aromatic ring-environmental pollutants, produced by the incomplete combustion of carbon-containing materials, such as coal, crude oil, wood, gasoline, food, and cigarettes [9]. Major sources of indoor PAH include emissions from residential heating, gas appliances, environmental tobacco smoke (ETS), and fumes from cooking, grilling, and frying. Previous studies have suggested a link between exposure to environmental pollutants such as PAH in concert with ETS postnatally and the onset of childhood respiratory symptoms or asthma [10,11]. Therefore, it is necessary to further explore and evaluate the association between prenatal PAH exposure and childhood-onset allergic diseases.
Allergic diseases are multifactorial, meaning that they result from a combination of environmental factors and genetic predisposition. This is illustrated by the rapid increase in the prevalence of allergic diseases in recent decades, which has accelerated too rapidly to be explained by genetic changes alone. The interaction between genetic predisposition and environmental exposure critically affects the developing immune system of neonates and children [8,12].
Genetic variations in glutathione S-transferases (GSTs) with different enzymatic activities can have a significant effect on children's susceptibility to atopic dermatitis [13]. The GST family comprises 3 superfamilies (cytosolic, mitochondrial, and microsomal) [14]. Human cytosolic GSTs belong to the alpha (GSTA1, GSTA2, GSTA3, GSTA4, and GSTA5), zeta (GSTZ1), theta (GSTT1, GSTT2, and GSTT4), mu (GSTM1, GSTM1L, GSTM2, GSTM3, GSTM4, and GSTM5), pi (GSTP1), sigma, and omega (GSTO1 and GSTO2) classes [15]. GSTs catalyze the conjugation of glutathione (GSH) to xenobiotic substrates to make them more water-soluble [14]. Their cellular defense activity against oxidative stress consists of detoxifying endogenous compounds such as peroxidized lipids and breaking down xenobiotics. GSTs may also bind with toxins and serve as transport proteins [16]. The general reaction of GST enzymes is the addition of GSH to electrophiles. There is a wide diversity of endogenous and exogenous electrophilic substrates, among which PAH are notable examples of exogenous electrophilic substrates [17]. GSTs catalyze the conjugation of GSH with PAH-derived free radicals. Previous studies have suggested the detoxifying properties of GSTs on ETS exposure [18,19], and have reported that children with prenatal exposure to ETS and GST deficiencies had a higher likelihood of asthma or decreased lung function.
It is difficult to identify the exact origin of allergic diseases because exposure factors interact with one another, and, in turn, these interactions affect allergic disease susceptibility. Therefore, this study sought to examine the effect of prenatal PAH exposure on allergic disease onset in 6-month-old infants. Among the various known GST isoenzymes, glutathione S-transferase M1 (GSTM1) and glutathione S-transferase T1 (GSTT1) are each encoded by different genes, and, if they are null allele variants, the null genotypes for both enzymes result in a lack of enzyme activity [20,21]. Therefore, we further assessed the association between prenatal PAH exposure and allergic diseases and how these relate to maternal GSTM1/GSTT1 polymorphisms to evaluate gene-environment interactions.
METHODS
This study was conducted as part of the Mothers and Children's Environmental Health (MOCEH) study. The MOCEH study is a multi-center birth cohort study with research centers in Seoul, Cheonan, and Ulsan in the Republic of Korea. Each center had a community-based network comprised of a university hospital and several public health centers. The MOCEH study was designed to investigate the effects of prenatal and postnatal pollutant exposures on growth, development, and health from fetal development to childhood. The pollutants assessed in the MOCEH cohort study were evaluated for endocrine disruptors, heavy metals, and PAH. The endocrine disruptors included bisphenol A and phthalate metabolites, and the harmful heavy metals included lead, mercury and cadmium, and the PAH included 1-hydroxypyrene (1-OHP) and 2-naphthol.
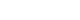
From 2006 to 2011, pregnant women living in the vicinity of the aforementioned locations, over the age of 18, and less than 20 weeks of gestational age were enrolled in the study along with their spouses (n = 1,751). In our study, pregnant women were selected based on whether information on both 1-OHP and 2-naphthol (n = 924) exposures were available. Furthermore, we excluded participants with < 30 mg/dL, > 300 mg/dL, or missing data on urinary creatinine (Cr) concentrations (n = 252). Those who did not respond to the self-administered food frequency questionnaire (FFQ) were also excluded (n = 50), and subjects with missing information regarding birth weight, gestational age, maternal body mass index (BMI), income, and maternal GSTM1/GSTT1 polymorphisms were excluded as per the complete case analysis (n = 18) [22]. Lastly, 6-month-old infants who had missing information on allergic diseases were also excluded (n = 255). In the final analysis, eligible participants for both the early period of pregnancy and late period of pregnancy (n = 349) were included in the study (Fig. 1).
Fig. 1
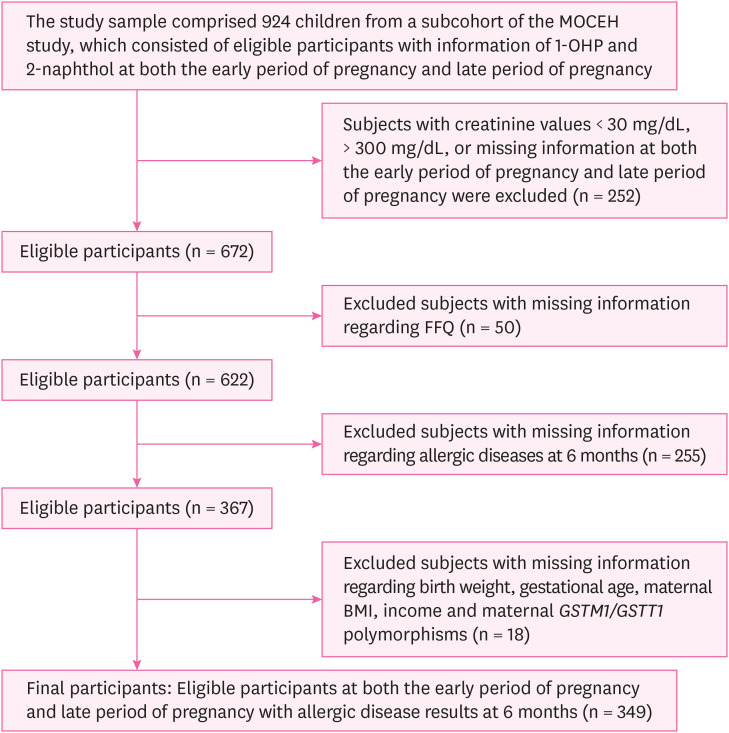
Flow diagram of the study design.
MOCEH: Mothers and Children's Environmental Health; 1-OHP: 1-hydroxypyrene; FFQ: food frequency questionnaire; BMI: body mass index; GSTM1: glutathione S-transferase M1; GSTT1: glutathione S-transferase T1.
Urinary 1-OHP and 2-naphthol levels were used as biomarkers of airborne PAH exposure [23]. Specifically, 1-OHP and 2-naphthol were measured in pregnant women via spot urine collections during their visits to the hospital throughout the early period of pregnancy (< 20 weeks of gestational age) and late period of pregnancy (> 28 weeks of gestational age). Maternal urine samples were collected in the morning and sent to a specialized laboratory for analysis. The 1-OHP and 2-naphthol urine concentrations were analyzed via high-performance liquid chromatography with fluorescence detection [24] and the levels measured during the early period of pregnancy and during late period of pregnancy were examined in the present study. The limit of detection (LOD) was 0.02 μg/L for 1-OHP and 0.057 μg/L for 2-naphthol, and samples below the LOD were divided by the square root of 2 [25].
Mothers/caregivers were asked about infantile symptoms, medical history, and the diagnosis of atopic dermatitis, asthma, food allergies, and any other allergic diseases or symptoms through an International Study of Asthma and Allergies in Childhood (ISAAC)-based questionnaire when the infants were 6 months old. ISAAC was established in 1991 and is the largest worldwide collaborative research project that focuses on asthma, rhinitis, and eczema in children. If an infant had symptoms, visited hospitals, or was diagnosed with either food allergies, atopic dermatitis, allergic rhinitis, allergic conjunctivitis, asthma, or other allergic symptoms, the infant was deemed to have an allergic disease [26].
Maternal DNA was extracted from maternal whole blood samples using the QIAamp DNA blood kit (Qiagen, Valencia, CA, USA). Polymerase chain reaction (PCR) was performed to identify maternal GSTM1 and GSTT1 polymorphisms. Moreover, a 268-bp β-globin gene fragment was amplified as a positive control. GSTM1 and GSTT1 genotyping were conducted with a PCR mixture containing 10 mM Tris-HCl, 40 mM KCl, 1.5 mM MgCl2, 0.25 mM of each dNTP, 1 unit of Taq polymerase (Bioneer, Seoul, Korea), 20 pmol of forward and reverse primers, and 50–100 ng of the genomic DNA as a template. The following GSTM1 and GSTT1 primer sets were used for PCR amplification: 5′-GAA CTC CCT GAA AAG CTA AAG C-3′ (forward) and 5′-GTT GGG CTC AAA TAT ACG GTG G-3′ (reverse) for GSTM1, and 5′-TCA CCG GAT CAT GGC CAG CA-3′ (forward) and 5′-TTC CTT ACT GGT CCT CAC ATC TC-3′ (reverse) for GSTT1. PCR amplification was performed with a PTC-200 thermal cycler (MJ Research, Watertown, MA, USA) with an initial denaturation at 94°C for 5 minutes; 35 cycles of denaturation at 94°C for 1 minutes, annealing at 65°C for 1 minutes, and extension at 72°C for 1 minutes; and a final extension at 72°C for 7 minutes. To examine the PCR-amplified fragments, electrophoresis was performed on a 3% 3:1 NuSieve/agarose gel (Cambrex Bio Science, Rockland, ME, USA). GSTM1 and GSTT1 genotyping were conducted based on the presence/absence of a 215-bp product and a 459-bp product, respectively. The GST null genotype was identified as a homozygous deletion of both of the examined genes. To confirm the results, 10% of the samples were randomly selected and analyzed once again to verify that the results were identical.
Trained nurses interviewed the study participants to collect information on maternal age, socioeconomic status, maternal education level, and parental allergic history using a structured questionnaire. Nutrition and dietary information were collected from the pregnant women during their prenatal visits using a semi-quantitative self-administered FFQ. The FFQ method has been previously validated in the Korean population [27]. The FFQ includes questions on the consumption frequency of beef, pork, and chicken products prepared by grilling, roasting, or smoking. The potential consumption frequency responses were divided into nine categories: ‘never or rare,’ ‘once monthly,’ ‘2 or 3 times a month,’ ‘once or twice a week,’ ‘3 or 4 times a week,’ ‘5 or 6 times a week,’ ‘daily,’ ‘twice a day,’ and ‘3 times a day.’ For downstream analyses, the maternal patterns of barbecued, fried, roasted, or grilled beef, pork, and chicken product consumption were further classified into 3 categories: (1) ‘1 time a month,’ (2) ‘2–3 times a month,’ and (3) ‘1–6 times a week’. The pre-pregnancy BMI was calculated as the body weight in kilograms divided by the height in meters squared. The participants were then divided into 3 groups: underweight (BMI < 18 kg/m2), normal weight (BMI 18–25 kg/m2), and overweight (BMI ≥ 25 kg/m2). Information related to the infant's sex, birth weight, and gestational age was obtained from medical records.
Differences in study participant characteristics were determined using the t-test for continuous variables and the χ2 test for categorical variables. The maternal urinary 1-OHP and 2-naphthol levels were corrected for Cr concentrations and were converted to a logarithmic base e scale to obtain a normal distribution. Logistic regression analysis was conducted to study the association between urinary 1-OHP and 2-naphthol levels during the early period of pregnancy and during late period of pregnancy and allergic diseases in 6-month-old infants. The covariates used in this study were the following: infant's sex (female versus male; categorical variable), gestational age (completed weeks; continuous variable), birth weight (grams; continuous variable), maternal age at delivery (< 30, ≥ 30 years old; ordinal variable), pre-pregnancy BMI (< 18, 18–25, ≥ 25; ordinal variable), family income (< 2, ≥ 2 million KRW; ordinal variable), maternal urinary cotinine concentration (ng/mL; < 18: non-smoker, 18–50: secondhand smoker, ≥ 50: active smoker; ordinal variable), maternal allergic history (yes or no; categorical variable), paternal allergic history (yes or no; categorical variable), and intake frequency of barbecued, fried, roasted or grilled beef, chicken, and pork (3 categories, as described above; ordinal variable).
Furthermore, to identify the interactive effect of GSTs and the metabolites of maternal PAH on infantile allergic diseases, an interaction between 1-OHP or 2-naphthol levels and GSTs was estimated. Interaction on an additive scale was estimated via the Relative Excess Risk due to Interaction (RERI) [28]. An RERI value larger than 0 suggested an additive interaction. The following formula was used to calculate the RERI: RERI = ORA × ORB × ORAB − ORA − ORB + 1, where ORA is the odds ratio (OR) of exposure to prenatal PAH, ORB is the OR of carrying different GST polymorphisms, and ORAB is the OR of the interaction of prenatal PAH and GSTs. After identifying any interaction effects, the association between prenatal PAH exposure and infantile allergic diseases in relation to the maternal GSTM1/GSTT1 polymorphisms was assessed. Stratified logistic regression analysis based on the GST null/present genotypes was performed to evaluate the effect of prenatal PAH exposure on allergic diseases in 6-month-old infants. Statistical significance was defined as p-value < 0.05. Data were analyzed using SAS version 9.4 (SAS Institute Inc., Cary, North Carolina, USA).
Before enrollment, written informed consent was obtained from the pregnant women, both for themselves and on behalf of their children. All protocols were approved by the Institutional Review Board of Ewha Womans University Hospital (approval No. 12-07B-15), Dankook University Hospital (approval No. 2011-09-0340), and Ulsan University Hospital (approval No.06-29).
RESULTS
The general characteristics of the 6-month-old infants with/without allergic diseases and those of their parents are summarized in Table 1. Of the 349 infants studied herein, 141 (40.4%) were affected by at least one allergic disease. There were no significant differences in maternal age among infants with/without allergic diseases. However, significant differences were observed in maternal BMI, indicating that mothers of non-allergic infants tended to weigh more. Moreover, parents of infants with allergic diseases tended to have a history of allergic diseases themselves. Interestingly, there was no significant difference in barbecued, fried, roasted, or grilled meat intake and cotinine concentration between the tested groups.
Table 1
Comparison of general characteristics of participant 6-month-old infants by allergic diseases
The geometric means of 1-OHP and 2-naphthol levels were 0.53 μg/g Cr and 2.12 μg/g Cr at the early period of pregnancy and 0.54 μg/g Cr and 2.09 μg/g Cr at late period of pregnancy, respectively (Table 2). The 2017 Korean National Environmental Health Survey (KoNEHS) reported urinary 1-OHP and 2-naphthol geometric means of 0.15 μg/g Cr and 3.16 μg/g Cr, respectively. The 2013–2014 National Health and Nutrition Examination Survey reported urinary 1-OHP and 2-naphthol geometric means of 0.15 μg/g Cr and 4.98 μg/g Cr, respectively.
Table 2
Distribution of 1-OHP and 2-naphthol levels in the maternal urine with correction for creatinine concentration at the early period of pregnancy and late period of pregnancy
Logistic regression analysis indicated that an increase in urinary 1-OHP during the early period of pregnancy was associated with an increased risk of allergic disease in 6-month-old infants (Table 3). When the 1-OHP concentration in the early period of pregnancy increased by one log-transformed unit of 1-OHP/Cr, the risk of allergic disease in 6-month-old infants increased by 1.53 times in the crude model, albeit not significantly. However, in the adjusted model, when the 1-OHP concentration in the early period of pregnancy increased by one log-transformed unit of 1-OHP/Cr, the risk of allergic disease in 6-month-old infants increased significantly by 1.84 times. We did not find a significant association between prenatal 2-naphthol and allergic diseases in 6-month-old infants.
Table 3
Association between maternal urinary PAH metabolites and allergic diseases in 6-month-old infants
Values are presented as odds ratio (95% confidence interval). Unit of the PAH metabolites, log-transformed μg/g creatinine.
PAH: polycyclic aromatic hydrocarbons; 1-OHP: 1-hydroxypyrene.
aModel adjusted for maternal age, maternal body mass index, birth weight, gestational age, infant's sex, family income, maternal allergic history, paternal allergic history, frequency of barbecued, fried, roasted or grilling beef, pork, and chicken, cotinine level (ng/mL); bGestational age < 20 weeks; cGestational age > 28 weeks; dThe p-value < 0.05.
The RERI results provided evidence of a positive additive interaction between 1-OHP in the early period of pregnancy and the GSTT1 null genotype but it was not statistically significant (RERI = 2.61; 95% confidence interval [CI]: −6.02, 11.24) (Table 4). Nevertheless, stratification analysis of maternal GSTT1 (null/present) supported the association between allergic diseases and 1-OHP in the early period of pregnancy (Table 4). In the adjusted model, when the 1-OHP concentration in the early period of pregnancy increased by one log-transformed unit of 1-OHP/Cr, the risk of allergic disease in 6-month-old infants with a maternal GSTT1 null genotype increased significantly by 2.69 times. However, no significant association was observed in the stratified analysis of maternal GSTM1. Additionally, the association between maternal GST genotype and allergic disease in 6-month-old infants, except for PAH exposure is described in Supplementary Table 1. The OR of allergic disease increased when GSTT1 was null compared to when GSTT1 was present but it was not statistically significant.
Table 4
Association between maternal urinary PAH metabolites and allergic diseases in 6-month-old infants in relation to maternal GST genotypes
Unit of the PAH metabolites, log-transformed μg/g creatinine.
PAH: polycyclic aromatic hydrocarbons; RERI: Relative Excess Risk due to Interaction; 1-OHP: 1-hydroxypyrene; OR: odds ratio; CI: confidence interval; GST: glutathione S-transferase; GSTM1: glutathione S-transferase M1; GSTT1: glutathione S-transferase T1.
aModel adjusted for maternal age, maternal BMI, birth weight, gestational age, infant's sex, family income, maternal allergic history, paternal allergic history, frequency of barbecued, fried, roasted or grilling beef, pork, and chicken, cotinine level (ng/mL); bGestational age < 20 weeks; cGestational age > 28 weeks; dThe p-value < 0.05.
DISCUSSION
The present study showed that 1-OHP exposure during the early period of pregnancy was associated with a significantly increased risk of allergic diseases in 6-month-old infants. Particularly, the association was limited to infants with a maternal GSTT1 null genotype; however, the RERI was not statistically significant.
PAH may contribute to increased prevalence and morbidity of allergic diseases. PAH on diesel exhaust particles (DEPs) can promote the mediated detoxification of the cytochrome P450 family 1 A1 [8]. In vitro and in vivo studies have demonstrated that inhalation of PAH-DEP upregulated interleukin (IL)-4 and IL-8 production [29,30,31]. A study by Takenaka et al. [32] showed that the PAH extract (obtained from PAH-DEP) enhanced the immunoglobulin E (IgE) production in the B cells due to IL-4. PAH can also inhibit the differentiation of human monocytes into macrophages, and this inhibitory effect may contribute to the potent immunotoxicity of PAH [33].
The developing immune functions in fetuses may be susceptible to prenatal PAH exposure, which can lead to adverse effects later in life [34]. A study from Spain showed that prenatal exposure to PAH was associated with the risk of respiratory infections in children during their first year of life [35]. Zhang et al. [36] reported that PAH were detected in the umbilical cord and placentas; they suggested the possibility of PAH transfer from the mother to the fetus via the placenta. Maternal exposure to PAH and placental transport can lead to the formation of DNA adducts easily, thereby confirming the heightened fetal susceptibility to prenatal PAH exposure [37,38]. Fetal T cells begin to differentiate between 8 and 12 weeks of gestational age [39,40]. Mature T helper cells or cytotoxic T cells appear at approximately 12 weeks of gestational age; subsequently, by 16 weeks of gestational age, CD4 and CD8 are expressed in the fetal thymus in patterns similar to the postnatal thymus, suggesting that human T cell repertoire is established by the end of the first trimester. At a similar time (approximately 11 weeks of gestational age), the fetus starts to produce IgE [41].
Various studies have suggested that the risk of allergic disease is determined by prenatal exposure to pollutants or chemicals that modify the T helper cell phenotype in postnatal life [42,43]. Polarization of naive CD4+ T cells into TH2 cells that induce IL-4 tends to inhibit interferon (IFN)-γ transcription by methylation of the IFN-γ promoter [44,45]. A birth cohort study in the Czech Republic examined short-term associations of air pollution, including PAH exposure with lymphocyte immunophenotypes in cord blood, thereby indicating that average ambient PAH levels before birth were associated with reductions in T-lymphocyte phenotype fractions [46]. A study by Tang et al. [47] reported that prenatal exposure to PAH was associated with promoter methylation of an asthma-related gene, IFN-γ, but not with the promoter methylation of IL-4. In addition, a study by Kim et al. [48] found that maternal cytokine levels started to polarize toward TH2-dominant immune response beginning in the first trimester and that a marked TH2-biased responsiveness increased susceptibility to childhood wheezing and atopy at 3 years of age. This suggests that any maternal cytokine dysregulation may start during early pregnancy, and marked TH2-biased responsiveness may induce the development of wheezing and atopic dermatitis in the offspring. Hence, exposure to PAH, which may cross the placenta, in the early period of pregnancy can lead to both maternal and fetal cytokine dysregulation and cause postnatal allergic diseases in infants and children. The mechanisms of PAH on fetal immune development between the first and second trimester, and how they are affected by epigenetics, remain to be elucidated; however, these findings support the potential effect of prenatal exposure to pollutants including PAH in the early period of pregnancy on fetal immune development.
The PAH metabolism pathway involves phase I drug metabolism enzymes (cytochrome P450) and phase II enzymes (primarily GSTs). GSTs catalyze the conjugation of PAH or their reactive metabolites to GSH, thereby making them easily excretable. The GSTs are polymorphic, and genetic polymorphisms in GSTs have been associated with abnormal PAH metabolism. A study by Chen et al. [49] reported a decreased 1-OHP excretion rate in Chinese coke oven workers with the GSTT1 null genotype, indicating that GSTT1 might be an important enzyme in the metabolism of PAH. Human GSTT1 is predominantly expressed in kidney and liver [50] with some expression in the placenta [51]. The exact role of GSTT1 in fetal and maternal detoxification is little known, but a higher proportion of GSTT1 compared to other GSTs suggests that GSTT1 is important in placental detoxification. Xenobiotics in the maternal blood can cross the placenta and accumulate in fetal organs [52]. Furthermore, deficiencies in maternal GSTT1, which is needed to detoxify xenobiotics, may lead to higher levels of toxicant exposure in their offspring. Summarizing the results of previous studies, fetuses with maternal GSTT1 null genotype may be exposed to more PAH in utero. Particularly, exposure in the early period of pregnancy may increase the likelihood of allergic disease after birth. Therefore, we could conclude that the results of our study may be explained by the aforementioned mechanisms and supported by the results of previous studies. However, the RERI was not statistically significant between allergic diseases in 6-month-old infants and maternal urinary PAH metabolites in relation to maternal GST genotypes. Thus, we could not firmly conclude that there was a significant additive interaction between 1-OHP exposure in the early period of pregnancy and maternal GSTT1 null genotype. In addition, more research on biological plausibility is needed.
To our knowledge, this is the first study to investigate the link between in utero exposure to PAH, maternal GSTT1 genotypes, and allergic disease onset in infants. Many studies have demonstrated direct links between childhood allergic diseases and the children's own GST genes; however, no studies have identified the association between childhood allergic disorders and maternal GSTT1 genes.
Another strength of this study is that, it was based on a cohort design with well-collected clinical and environmental exposure data. Such a multi-generational study design has the potential to provide the required resolution for the examination of early life disease risk factors. Furthermore, since this study was based on a prospective cohort study, it enabled us to limit recall bias. The main advantage of this study was that the effects of prenatal PAH exposure were studied during both the early period of pregnancy and late period of pregnancy. Therefore, our study identified the critical window of prenatal PAH exposure associated with allergic disease risk in 6-month-old infants.
The main limitation of the present study was that the effect of prenatal PAH exposure could not be clearly distinguished from postnatal environmental exposure. However, we addressed this limitation by adjusting the frequency of barbecued, fried, roasted, or grilled beef, pork, and chicken and the concentration of maternal cotinine. Given the early age of the children involved in this study (6 months), neither skin prick tests nor pulmonary function tests could be performed.
Another limitation was that urine measurement of 1-OHP and 2-naphthol at the early period of pregnancy or late period of pregnancy might not reflect the whole exposure during the early period of pregnancy or late period of pregnancy due to the short half-life of 1-OHP and 2-naphthol. In humans, the urinary elimination rate of 1-OHP varies considerably depending on the exposure routes. The half-life of urinary 1-OHP varies from 3.0 to 5.7 hours, 3.7 to 9.9 hours, and 11.5 to 15.0 hours after oral, inhalation, and dermal absorption, respectively [53]. It could have been influenced by smoking, exposure to exhaust gas, or the diet on the day before the measurement. To compensate for this limitation, the cotinine concentration was adjusted in the analysis; however, this method could not have completely ruled out the effects of dietary intake or exhaust gas exposure on the previous day. In addition, when the maternal urine samples were collected, they were not collected at defined weeks of pregnancy and instead they were sampled at various times over the period of the early or late pregnancy. This could affect the interpretation of the results.
Also, we did not find a significant additive interaction between 1-OHP exposure in the early period of pregnancy and a maternal GSTT1 null genotype. It is acknowledged that studies on gene-environmental interaction require a sample size at least 4 times larger than one required to detect the main effect of the same magnitude [54]. Thus, due to the small sample size of our study, we could not get significant interaction effects in relation to maternal GST genotypes.
Lastly, the final study participants accounted for only 37.8% (n = 349) of the initial study population, and this might have led to selection bias. Therefore, we compared the general characteristics of the participants that were included in the analysis versus those of the excluded participants (Supplementary Table 2). We found that there were no statistically significant differences between the included and excluded participants and therefore, selection bias could be ruled out.
CONCLUSION
Our study showed the effect of early prenatal exposure to PAH on the likelihood of allergic disease onset in infants and how this relates to maternal GSTT1 polymorphism. However, the underlying mechanisms of these associations still require further exploration in future studies.
Abbreviations
BMI
body mass index
CI
confidence interval
Cr
creatinine
DEP
diesel exhaust particle
ETS
environmental tobacco smoke
FFQ
food frequency questionnaire
GM
geometric mean
GSD
geometric standard deviation
GSH
glutathione
GST
glutathione S-transferase
GSTM1
glutathione S-transferase M1
GSTT1
glutathione S-transferase T1
IFN
interferon
IgE
immunoglobulin E
IL
interleukin
ISAAC
International Study of Asthma and Allergies in Childhood
LOD
limit of detection
Min.
Minimum
Max.
Maximum
MOCEH
Mothers and Children's Environmental Health
OR
odds ratio
PAH
polycyclic aromatic hydrocarbons
PCR
polymerase chain reaction
RERI
Relative Excess Risk due to Interaction
1-OHP
1-hydroxypyrene
-
Funding: This study was supported by the MOCEH (Mothers and Children's Environmental Health) project of the National Institute of Environmental Research, Republic of Korea. This work was supported by a grant from the National Institute of Environment Research (NIER), funded by the Ministry of Environment (MOE) of the Republic of Korea (NIER-2012-00-02-089).
-
Competing interests: The authors declare that they have no competing interests.
-
Author Contributions:
Conceptualization: Koh TK, Shah S, Ha E.
Data curation: Koh TK, Shah S.
Formal analysis: Koh TK, Shah S.
Methodology: Koh TK, Shah S, Ha E.
Project administration: Koh TK, Shah S, Ha E.
Software: Koh TK, Shah S.
Writing - original draft: Koh TK, Shah S.
Writing - review & editing: Koh TK, Shah S, Ha E, Park H, Hong YC, Ha M, Kim Y, Lee BE.
NOTES
SUPPLEMENTARY MATERIALS
Supplementary Table 1
Association between maternal GST genotypes and allergic diseases in 6-month-old infants
Supplementary Table 2
Characteristics of study participants who were included in our analyses versus excluded
- 1. Mota IA, Borrego LM. Allergic response to fungal exposure. In: Viegas C, Pinheiro AC, Sabino R, Viegas S, Brandão J, Veríssimo C, editors. Environmental Mycology in Public Health. Cambridge, MA: Academic Press; 2016, 35–43.
- 2. Silverberg JI, Simpson EL. Association between severe eczema in children and multiple comorbid conditions and increased healthcare utilization. Pediatr Allergy Immunol 2013;24(5):476–486. 23773154.ArticlePubMedPMC
- 3. Adnan C. Epidemiology of allergic diseases. In: Robyn EO, Stephen TH, Aziz S, editors. Middleton's Allergy Essentials. Amsterdam: Elsevier; 2017, 57–60.
- 4. Kim BK, Kim JY, Kang MK, Yang MS, Park HW, Min KU, et al. Allergies are still on the rise? A 6-year nationwide population-based study in Korea. Allergol Int 2016;65(2):186–191. 26666496.ArticlePubMed
- 5. Lee E, Lee SY, Yang HJ, Hong SJ. Epidemiology of allergic diseases in Korean children. Allergy Asthma Respir Dis 2018;6(Suppl 1):S9–S20.ArticlePDF
- 6. Yoon SJ. A Study on Research Methodology and Long-term Planning Regarding Estimation of Economic Burden of Major Diseases in Korea. Seoul: Korea University; 2009.
- 7. Gluckman P, Buklijas T, Hanson M. The Epigenome and Developmental Origins of Health and Disease. 1st ed. Cambridge, MA: Academic Press; 2016.
- 8. Murrison LB, Brandt EB, Myers JB, Hershey GK. Environmental exposures and mechanisms in allergy and asthma development. J Clin Invest 2019;129(4):1504–1515. 30741719.ArticlePubMedPMC
- 9. Menichini E, Bocca B. Polycyclic aromatic hydrocarbons. In: Caballero B, editor. Encyclopedia of Food Sciences and Nutrition. Cambridge, MA: Academic Press; 2003, 4616–4625.
- 10. Miller RL, Garfinkel R, Horton M, Camann D, Perera FP, Whyatt RM, et al. Polycyclic aromatic hydrocarbons, environmental tobacco smoke, and respiratory symptoms in an inner-city birth cohort. Chest 2004;126(4):1071–1078. 15486366.ArticlePubMed
- 11. Rosa MJ, Jung KH, Perzanowski MS, Kelvin EA, Darling KW, Camann DE, et al. Prenatal exposure to polycyclic aromatic hydrocarbons, environmental tobacco smoke and asthma. Respir Med 2011;105(6):869–876. 21163637.ArticlePubMedPMC
- 12. Kim BJ, Lee SY, Kim HB, Lee E, Hong SJ. Environmental changes, microbiota, and allergic diseases. Allergy Asthma Immunol Res 2014;6(5):389–400. 25228995.ArticlePubMedPMC
- 13. Wen HJ, Wang SL, Chen PC, Guo YL. Prenatal perfluorooctanoic acid exposure and glutathione s-transferase T1/M1 genotypes and their association with atopic dermatitis at 2 years of age. PLoS One 2019;14(1):e0210708. 30650146.ArticlePubMedPMC
- 14. Oakley A. Glutathione transferases: a structural perspective. Drug Metab Rev 2011;43(2):138–151. 21428697.ArticlePubMed
- 15. Idowu AT, Mujeeb SO. Polymorphic human glutathione s-transferase genes may predict susceptibility to type 2 diabetes mellitus: a minireview. Int J Biomed Res 2015;6(3):139–143.ArticlePDF
- 16. Leaver MJ, George SG. A piscine glutathione S-transferase which efficiently conjugates the end-products of lipid peroxidation. Mar Environ Res 1998;46(1-5):71–74.Article
- 17. Strange RC, Spiteri MA, Ramachandran S, Fryer AA. Glutathione-S-transferase family of enzymes. Mutat Res 2001;482(1-2):21–26. 11535245.ArticlePubMed
- 18. Breton CV, Vora H, Salam MT, Islam T, Wenten M, Gauderman WJ, et al. Variation in the GST mu locus and tobacco smoke exposure as determinants of childhood lung function. Am J Respir Crit Care Med 2009;179(7):601–607. 19151192.ArticlePubMedPMCPDF
- 19. Kabesch M, Hoefler C, Carr D, Leupold W, Weiland SK, von Mutius E. Glutathione S transferase deficiency and passive smoking increase childhood asthma. Thorax 2004;59(7):569–573. 15223862.ArticlePubMedPMC
- 20. Pemble S, Schroeder KR, Spencer SR, Meyer DJ, Hallier E, Bolt HM, et al. Human glutathione S-transferase theta (GSTT1): cDNA cloning and the characterization of a genetic polymorphism. Biochem J 1994;300(Pt 1):271–276. 8198545.ArticlePubMedPMCPDF
- 21. Brockmöller J, Gross D, Kerb R, Drakoulis N, Roots I. Correlation between trans-stilbene oxide-glutathione conjugation activity and the deletion mutation in the glutathione S-transferase class mu gene detected by polymerase chain reaction. Biochem Pharmacol 1992;43(3):647–650. 1540219.ArticlePubMed
- 22. Langkamp DL, Lehman A, Lemeshow S. Techniques for handling missing data in secondary analyses of large surveys. Acad Pediatr 2010;10(3):205–210. 20338836.ArticlePubMedPMC
- 23. Kim H, Cho SH, Kang JW, Kim YD, Nan HM, Lee CH, et al. Urinary 1-hydroxypyrene and 2-naphthol concentrations in male Koreans. Int Arch Occup Environ Health 2001;74(1):59–62. 11196083.ArticlePubMedPDF
- 24. Bae S, Pan XC, Kim SY, Park K, Kim YH, Kim H, et al. Exposures to particulate matter and polycyclic aromatic hydrocarbons and oxidative stress in schoolchildren. Environ Health Perspect 2010;118(4):579–583. 20368125.ArticlePubMed
- 25. Hornung RW, Reed LD. Estimation of average concentration in the presence of nondetectable values. Appl Occup Environ Hyg 1990;5(1):46–51.Article
- 26. Okada E, Sasaki S, Kashino I, Matsuura H, Miyashita C, Kobayashi S, et al. Prenatal exposure to perfluoroalkyl acids and allergic diseases in early childhood. Environ Int 2014;65:127–134. 24486970.ArticlePubMed
- 27. Kim DW, Song S, Lee JE, Oh K, Shim J, Kweon S, et al. Reproducibility and validity of an FFQ developed for the Korea National Health and Nutrition Examination Survey (KNHANES). Public Health Nutr 2015;18(8):1369–1377. 25167205.ArticlePubMedPMC
- 28. Knol MJ, van der Tweel I, Grobbee DE, Numans ME, Geerlings MI. Estimating interaction on an additive scale between continuous determinants in a logistic regression model. Int J Epidemiol 2007;36(5):1111–1118. 17726040.ArticlePubMed
- 29. Devouassoux G, Saxon A, Metcalfe DD, Prussin C, Colomb MG, Brambilla C, et al. Chemical constituents of diesel exhaust particles induce IL-4 production and histamine release by human basophils. J Allergy Clin Immunol 2002;109(5):847–853. 11994710.ArticlePubMed
- 30. Salvi SS, Nordenhall C, Blomberg A, Rudell B, Pourazar J, Kelly FJ, et al. Acute exposure to diesel exhaust increases IL-8 and GRO-α production in healthy human airways. Am J Respir Crit Care Med 2000;161(2 Pt 1):550–557. 10673199.ArticlePubMedPDF
- 31. Bömmel H, Li-Weber M, Serfling E, Duschl A. The environmental pollutant pyrene induces the production of IL-4. J Allergy Clin Immunol 2000;105(4):796–802. 10756232.ArticlePubMed
- 32. Takenaka H, Zhang K, Diaz-Sanchez D, Tsien A, Saxon A. Enhanced human IgE production results from exposure to the aromatic hydrocarbons from diesel exhaust: direct effects on B-cell IgE production. J Allergy Clin Immunol 1995;95(1 Pt 1):103–115. 7529782.ArticlePubMed
- 33. van Grevenynghe J, Rion S, Le Ferrec E, Le Vee M, Amiot L, Fauchet R, et al. Polycyclic aromatic hydrocarbons inhibit differentiation of human monocytes into macrophages. J Immunol 2003;170(5):2374–2381. 12594260.ArticlePubMedPDF
- 34. Jedrychowski W, Galas A, Pac A, Flak E, Camman D, Rauh V, et al. Prenatal ambient air exposure to polycyclic aromatic hydrocarbons and the occurrence of respiratory symptoms over the first year of life. Eur J Epidemiol 2005;20(9):775–782. 16170661.ArticlePubMedPDF
- 35. Jerzynska J, Podlecka D, Polanska K, Hanke W, Stelmach I, Stelmach W. Prenatal and postnatal exposure to polycyclic aromatic hydrocarbons and allergy symptoms in city children. Allergol Immunopathol (Madr) 2017;45(1):18–24. 27789067.ArticlePubMed
- 36. Zhang X, Li X, Jing Y, Fang X, Zhang X, Lei B, et al. Transplacental transfer of polycyclic aromatic hydrocarbons in paired samples of maternal serum, umbilical cord serum, and placenta in Shanghai, China. Environ Pollut 2017;222:267–275. 28024810.ArticlePubMed
- 37. Perera F, Tang D, Whyatt R, Lederman SA, Jedrychowski W. DNA damage from polycyclic aromatic hydrocarbons measured by benzo[a]pyrene-DNA adducts in mothers and newborns from Northern Manhattan, the World Trade Center Area, Poland, and China. Cancer Epidemiol Biomarkers Prev 2005;14(3):709–714. 15767354.ArticlePubMedPDF
- 38. Jedrychowski WA, Perera FP, Tang D, Rauh V, Majewska R, Mroz E, et al. The relationship between prenatal exposure to airborne polycyclic aromatic hydrocarbons (PAHs) and PAH-DNA adducts in cord blood. J Expo Sci Environ Epidemiol 2013;23(4):371–377. 23299301.ArticlePubMedPMCPDF
- 39. Haynes BF, Heinly CS. Early human T cell development: analysis of the human thymus at the time of initial entry of hematopoietic stem cells into the fetal thymic microenvironment. J Exp Med 1995;181(4):1445–1458. 7699329.ArticlePubMedPMCPDF
- 40. Lobach DF, Hensley LL, Ho W, Haynes BF. Human T cell antigen expression during the early stages of fetal thymic maturation. J Immunol 1985;135(3):1752–1759. 3926883.ArticlePubMedPDF
- 41. Miller DL, Hiravonen T, Gitlin D. Synthesis of IgE by the human conceptus. J Allergy Clin Immunol 1973;52(3):182–188. 4737557.ArticlePubMed
- 42. McFadden JP, Thyssen JP, Basketter DA, Puangpet P, Kimber I. T helper cell 2 immune skewing in pregnancy/early life: chemical exposure and the development of atopic disease and allergy. Br J Dermatol 2015;172(3):584–591. 25354210.ArticlePubMed
- 43. Miller RL, Ho SM. Environmental epigenetics and asthma: current concepts and call for studies. Am J Respir Crit Care Med 2008;177(6):567–573. 18187692.PubMedPMC
- 44. Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol 2002;2(12):933–944. 12461566.ArticlePubMedPDF
- 45. Jones B, Chen J. Inhibition of IFN-gamma transcription by site-specific methylation during T helper cell development. EMBO J 2006;25(11):2443–2452. 16724115.ArticlePubMedPMCPDF
- 46. Hertz-Picciotto I, Herr CE, Yap PS, Dostál M, Shumway RH, Ashwood P, et al. Air pollution and lymphocyte phenotype proportions in cord blood. Environ Health Perspect 2005;113(10):1391–1398. 16203253.ArticlePubMedPMC
- 47. Tang WY, Levin L, Talaska G, Cheung YY, Herbstman J, Tang D, et al. Maternal exposure to polycyclic aromatic hydrocarbons and 5′-CpG methylation of interferon-γ in cord white blood cells. Environ Health Perspect 2012;120(8):1195–1200. 22562770.ArticlePubMedPMC
- 48. Kim JH, Kim KH, Woo HY, Shim JY. Maternal cytokine production during pregnancy and the development of childhood wheezing and allergic disease in offspring three years of age. J Asthma 2008;45(10):948–952. 19085588.PubMed
- 49. Chen B, Hu Y, Jin T, Lu D, Shao M, Zheng L, et al. The influence of metabolic gene polymorphisms on urinary 1-hydroxypyrene concentrations in Chinese coke oven workers. Sci Total Environ 2007;381(1-3):38–46. 17498780.ArticlePubMed
- 50. Hayes JD, Strange RC. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology 2000;61(3):154–166. 10971201.ArticlePubMedPDF
- 51. Raijmakers MT, Bruggeman SW, Steegers EA, Peters WH. Distribution of components of the glutathione detoxification system across the human placenta after uncomplicated vaginal deliveries. Placenta 2002;23(6):490–496. 12137747.ArticlePubMed
- 52. Krauer B, Dayer P. Fetal drug metabolism and its possible clinical implications. Clin Pharmacokinet 1991;21(1):70–80. 1914342.ArticlePubMed
- 53. Lutier S, Maître A, Bonneterre V, Bicout DJ, Marques M, Persoons R, et al. Urinary elimination kinetics of 3-hydroxybenzo(a)pyrene and 1-hydroxypyrene of workers in a prebake aluminum electrode production plant: evaluation of diuresis correction methods for routine biological monitoring. Environ Res 2016;147:469–479. 26970901.ArticlePubMed
- 54. Smith PG, Day NE. The design of case-control studies: the influence of confounding and interaction effects. Int J Epidemiol 1984;13(3):356–365. 6386716.ArticlePubMed
REFERENCES
REFERENCES
Figure & Data
REFERENCES
Citations
Citations to this article as recorded by 
- Environmental Pollutants and Food Allergy: From Traditional Triggers to Emerging Hazards
Qiang Shi, Xiang Gao, Shan Zhang, Yuhong Wu, Huan Wu, Haojie Luo, Yanhai Xie, Shuangyan Zheng, Yong Wu, Xin Li, Jinyan Gao, Jinlyu Sun, Zhongliang Wang, Hongbing Chen
Clinical Reviews in Allergy & Immunology.2026;[Epub] CrossRef - Infantile allergic diseases: a cohort study prenatal fish intake and mercury exposure context
Surabhi Shah, Hae Soon Kim, Yun-Chul Hong, Hyesook Park, Mina Ha, Yangho Kim, Ji Hyen Lee, Eun-Hee Ha
BMC Public Health.2024;[Epub] CrossRef - Prenatal dietary exposure to mixtures of chemicals is associated with allergy or respiratory diseases in children in the ELFE nationwide cohort
Manel Ghozal, Manik Kadawathagedara, Rosalie Delvert, Amandine Divaret-Chauveau, Chantal Raherison, Raphaëlle Varraso, Annabelle Bédard, Amélie Crépet, Véronique Sirot, Marie Aline Charles, Karine Adel-Patient, Blandine de Lauzon-Guillain
Environmental Health.2024;[Epub] CrossRef - Prenatal dietary exposure to chemicals and allergy or respiratory diseases in children in the EDEN mother–child cohort
Manel Ghozal, Manik Kadawathagedara, Rosalie Delvert, Karine Adel-Patient, Muriel Tafflet, Isabella Annesi-Maesano, Amélie Crépet, Véronique Sirot, Marie Aline Charles, Barbara Heude, Blandine de Lauzon-Guillain
Environment International.2023; 180: 108195. CrossRef - Gene-environment interactions related to maternal exposure to environmental and lifestyle-related chemicals during pregnancy and the resulting adverse fetal growth: a review
Sumitaka Kobayashi, Fumihiro Sata, Reiko Kishi
Environmental Health and Preventive Medicine.2022; 27: 24. CrossRef - Role of GSTM1 in Hypertension, CKD, and Related Diseases across the Life Span
Rebecca Levy, Thu H. Le
Kidney360.2022; 3(12): 2153. CrossRef
 Cite
CiteAssociation between prenatal polycyclic aromatic hydrocarbons and infantile allergic diseases modified by maternal glutathione S-transferase polymorphisms: results from the MOCEH birth cohort
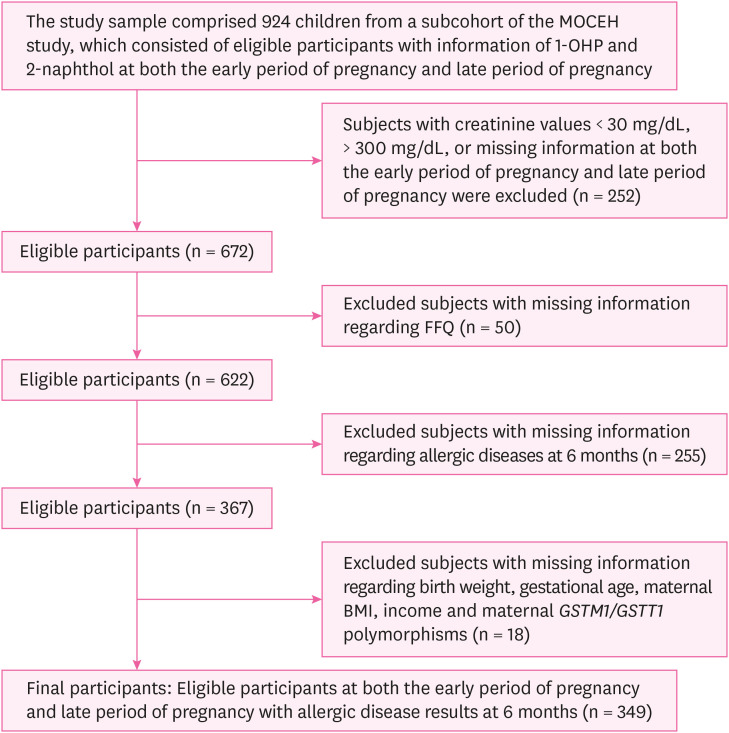
Fig. 1 Flow diagram of the study design.MOCEH: Mothers and Children's Environmental Health; 1-OHP: 1-hydroxypyrene; FFQ: food frequency questionnaire; BMI: body mass index; GSTM1: glutathione S-transferase M1; GSTT1: glutathione S-transferase T1.
Fig. 1
Association between prenatal polycyclic aromatic hydrocarbons and infantile allergic diseases modified by maternal glutathione S-transferase polymorphisms: results from the MOCEH birth cohort
| Variables | Total | Not having allergic diseases | Having allergic diseases | p-valuea | |
|---|---|---|---|---|---|
| Total | 349 (100.0) | 208 (59.6) | 141 (40.4) | < 0.01c | |
| Sex of the child | 0.86 | ||||
| Male | 191 (54.7) | 113 (54.3) | 78 (55.3) | ||
| Female | 158 (45.3) | 95 (45.7) | 63 (44.7) | ||
| Maternal age (year) | 0.06 | ||||
| < 30 | 164 (47.0) | 89 (42.8) | 75 (53.2) | ||
| ≥ 30 | 185 (53.0) | 119 (57.2) | 66 (46.8) | ||
| Maternal BMI (kg/m2) | 0.05c | ||||
| 0 < BMI <18 | 5 (1.4) | 3 (1.4) | 2 (1.4) | ||
| 18.0 ≤ BMI < 25.0 | 259 (74.2) | 145 (69.7) | 114 (80.9) | ||
| BMI ≥ 25.0 | 85 (24.4) | 60 (28.9) | 25 (17.7) | ||
| Income (million KRW/month) | 0.79 | ||||
| < 2 | 89 (25.5) | 52 (25.0) | 37 (26.2) | ||
| ≥ 2 | 260 (74.5) | 156 (75.0) | 104 (73.8) | ||
| Maternal allergic disease history | 0.01c | ||||
| No | 234 (67.1) | 150 (72.1) | 84 (59.6) | ||
| Yes | 115 (32.9) | 58 (27.9) | 57 (40.4) | ||
| Paternal allergic disease history | 0.04c | ||||
| No | 255 (73.1) | 160 (76.9) | 95 (67.4) | ||
| Yes | 94 (26.9) | 48 (23.1) | 46 (32.6) | ||
| Barbecued/fried beefb | 0.96 | ||||
| 1 time/month | 181 (51.9) | 108 (51.9) | 73 (51.8) | ||
| 2–3 times/month | 106 (30.4) | 64 (30.8) | 42 (29.8) | ||
| 1–6 times/week | 62 (17.8) | 36 (17.3) | 26 (18.4) | ||
| Barbecued/fried porkb | 0.93 | ||||
| 1 time/month | 37 (10.6) | 21 (10.1) | 16 (11.4) | ||
| 2–3 times/month | 120 (34.4) | 72 (34.6) | 48 (34.0) | ||
| 1–6 times/week | 192 (55.0) | 115 (55.3) | 77 (54.6) | ||
| Barbecued/fried chickenb | 0.35 | ||||
| 1 time/month | 122 (35.0) | 79 (38.0) | 43 (30.5) | ||
| 2–3 times/month | 138 (39.5) | 79 (38.0) | 59 (41.8) | ||
| 1–6 times/week | 89 (25.5) | 50 (24.0) | 39 (27.7) | ||
| Cotinine at early period of pregnancy (ng/mL) | 0.75 | ||||
| 0 < Cotinine < 18 | 333 (95.4) | 197 (94.7) | 136 (96.5) | ||
| 18 ≤ Cotinine < 50 | 3 (0.9) | 2 (1.0) | 1 (0.7) | ||
| Cotinine ≥ 50 | 13 (3.7) | 9 (4.3) | 4 (2.8) | ||
| Cotinine at late period of pregnancy (ng/mL) | 0.41 | ||||
| 0 < Cotinine < 18 | 334 (95.7) | 197 (94.7) | 137 (97.2) | ||
| 18 ≤ Cotinine < 50 | 5 (1.4) | 3 (1.4) | 2 (1.4) | ||
| Cotinine ≥ 50 | 10 (2.9) | 8 (3.9) | 2 (1.4) | ||
| GSTM1 | 0.57 | ||||
| Null | 192 (55.0) | 117 (56.3) | 75 (53.2) | ||
| Positive | 157 (45.0) | 91 (43.7) | 66 (46.8) | ||
| GSTT1 | 0.13 | ||||
| Null | 176 (50.4) | 98 (47.1) | 78 (55.3) | ||
| Positive | 173 (49.6) | 110 (52.9) | 63 (44.7) | ||
| PAH metabolites | Gestation (n = 349) | GM | GSD | Min | 25th percentiles | 50th percentiles | 75th percentiles | Max |
|---|---|---|---|---|---|---|---|---|
| 1-OHP (μg/g creatinine) | Earlya | 0.53 | 1.52 | 0.13 | 0.39 | 0.54 | 0.73 | 1.39 |
| Lateb | 0.54 | 1.49 | 0.16 | 0.42 | 0.55 | 0.73 | 1.94 | |
| 2-naphthol (μg/g creatinine) | Earlya | 2.12 | 1.63 | 0.28 | 1.56 | 2.21 | 2.85 | 7.35 |
| Lateb | 2.09 | 1.59 | 0.23 | 1.54 | 2.15 | 2.77 | 9.23 |
| PAH metabolites | Risk of allergic diseases in 6-month-old infants (n = 349) | ||
|---|---|---|---|
| Crude | Adjusteda | ||
| Early period of pregnancyb | |||
| 1-OHP | 1.53 (0.91, 2.58) | 1.84 (1.05, 3.22)d | |
| 2-naphthol | 1.08 (0.70, 1.68) | 1.03 (0.64, 1.65) | |
| Late period of pregnancyc | |||
| 1-OHP | 1.40 (0.81, 2.40) | 1.50 (0.83, 2.71) | |
| 2-naphthol | 0.98 (0.62, 1.56) | 0.97 (0.59, 1.61) | |
| Risk of allergic diseases in 6-month-old infantsa | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Number (%) | Early period of pregnancyb | Late period of pregnancyc | |||||||
| 1-OHP | 2-naphthol | 1-OHP | 2-naphthol | |||||||
| OR (95% CI)a | RERI (95% CI) | OR (95% CI)a | RERI (95% CI) | OR (95% CI)a | RERI (95% CI) | OR (95% CI)a | RERI (95% CI) | |||
| GSTT1 | ||||||||||
| Null | 176 (50.4) | 2.69 (1.17, 6.21)d | 2.61 (−6.02, 11.24) | 0.68 (0.33, 1.39) | −0.25 (−0.53, 0.04) | 2.37 (0.96, 5.83) | 3.62 (−7.79, 15.04) | 1.53 (0.66, 3.52) | 0.44 (−0.99, 1.88) | |
| Present | 173 (49.6) | 1.36 (0.58, 3.20) | 1.50 (0.70, 3.21) | 0.93 (0.40, 2.16) | 0.64 (0.31, 1.33) | |||||
| GSTM1 | ||||||||||
| Null | 192 (55.0) | 1.16 (0.50, 2.67) | −0.78 (−1.67, 0.11) | 0.92 (0.48, 1.76) | −0.12 (−0.52, 0.28) | 1.49 (0.65, 3.42) | 0.23 (−1.98, 2.44) | 0.97 (0.43, 2.20) | 0.01 (−0.45, 0.46) | |
| Present | 157 (45.0) | 2.23 (0.83, 5.20) | 1.31 (0.59, 2.91) | 1.35 (0.52, 3.48) | 1.09 (0.54, 2.22) | |||||
Table 1 Comparison of general characteristics of participant 6-month-old infants by allergic diseases
Values are presented as number (%).
BMI: body mass index;
aTested by χ2 or Fisher's exact test; bObtained from the food frequency questionnaire; cThe
Table 2 Distribution of 1-OHP and 2-naphthol levels in the maternal urine with correction for creatinine concentration at the early period of pregnancy and late period of pregnancy
PAH: polycyclic aromatic hydrocarbons; 1-OHP: 1-hydroxypyrene; GM: geometric mean; GSD: geometric standard deviation; Min.: Minimum; Max.: Maximum.
aGestational age < 20 weeks; bGestational age > 28 weeks.
Table 3 Association between maternal urinary PAH metabolites and allergic diseases in 6-month-old infants
Values are presented as odds ratio (95% confidence interval). Unit of the PAH metabolites, log-transformed μg/g creatinine.
PAH: polycyclic aromatic hydrocarbons; 1-OHP: 1-hydroxypyrene.
aModel adjusted for maternal age, maternal body mass index, birth weight, gestational age, infant's sex, family income, maternal allergic history, paternal allergic history, frequency of barbecued, fried, roasted or grilling beef, pork, and chicken, cotinine level (ng/mL); bGestational age < 20 weeks; cGestational age > 28 weeks; dThe
Table 4 Association between maternal urinary PAH metabolites and allergic diseases in 6-month-old infants in relation to maternal GST genotypes
Unit of the PAH metabolites, log-transformed μg/g creatinine.
PAH: polycyclic aromatic hydrocarbons; RERI: Relative Excess Risk due to Interaction; 1-OHP: 1-hydroxypyrene; OR: odds ratio; CI: confidence interval; GST: glutathione S-transferase;
aModel adjusted for maternal age, maternal BMI, birth weight, gestational age, infant's sex, family income, maternal allergic history, paternal allergic history, frequency of barbecued, fried, roasted or grilling beef, pork, and chicken, cotinine level (ng/mL); bGestational age < 20 weeks; cGestational age > 28 weeks; dThe